




物性研(System B Ohtaka)でのQuantum Espresso-Perturboコンパイル
これも内輪用です!(重要!)
責任は取れないよ!
下準備
VASPのコンパイルの時と下準備は一緒です。
HDF5は既にインストール済みでしたら適宜スキップしてください。
まずはmodule loadから
module purge module load oneapi_compiler/2023.0.0 oneapi_mkl/2023.0.0 oneapi_mpi/2023.0.0
次にHDF5のインストール
HDF5® Source Code - The HDF Group
ここからソースコードをダウンロードしてください。
ダウンロードできたらHDF5のディレクトリ内で以下を実行します。
prefix=は各々のディレクトリを指定してください。
./configure --prefix=/home/test/hdf5-1.8.18/ --enable-fortran CC=mpicc FC=mpiifort CXX=mpicxx make make install
Quantum Espressoのコンパイル
Quantum Espressoのバージョンは7.2を使用しています。
Quantum Espresso(QE)のディレクトリ直下でconfigureを実行します。
.configure
実行したら"make.inc"ができると思うので一応中身を確認しておきましょう。
module loadは絶対に忘れずにね!
もしHDF5をQEでも使うのなら
# HDF5 HDF_LIBS = /your/hdf5/lib
って感じで追記しておきましょう。
そして各自使うやつをmakeしましょう。
make pw ph pp w90
makeする時はジョブスクリプト書くかインタラクティブジョブ状態にしてからしようね!
約束だよ!(超重要!)
QEのコンパイルはこれで完了です。
./binの中に"~.x"といった感じの実行ファイルができているはずです。
Wannier90のコンパイル
ここからダウンロードできます。
PerturboはWannier 90-3.0.0以降しか対応していないのでそこだけ注意。
"config"の中にある"make.inc.~"をコピーします。(ここではmake.inc.ifort)
cp ./config/make.inc.ifort ./make.inc
そしてmakeすれば難なくコンパイルできるはずです。
Perturbo
Perturboのバージョンは2.1.0を使用しています。
バージョン2.1.0の場合、ディレクトリ直下に"make.sys"があるのでそれを編集していきます。
ちなみに変更する箇所が多くなりすぎるので、PerturboはQEのディレクトリ内に置くことを強く勧めます。
編集といっても通常はHDFのPATHを書いてあげるだけでOKです。
#path to HDF5 library IFLAGS += -I/home/k~/k~/hdf5~/include HDF5_LIB = -L/home/k~/k~/hdf5/lib -lhdf5 -lhdf5_fortran
書いてあげたらmakeして、"bin"ディレクトリ内に"perturbo.x"と"qe2pert.x"があれば大丈夫です。
おまけ
濱田先生のQiitaに書いてある通り
ジョブスクリプトに
export FI_PROVIDER=psm3
を追記してあげた方が実行速度が速くなっている気がします。
ohtaka上でのIntelコンパイラ(+IntelMPI)を使ったコンパイルの問題 #IntelCompiler - Qiita
参考までに自分のジョブスクリプトはこんな感じです。
~~~省略~~~ ulimit -s unlimited module purge module load oneapi_compiler/2023.0.0 oneapi_mkl/2023.0.0 oneapi_mpi/2023.0.0 export MKL_DEBUG_CPU_TYPE=5 export UCX_TLS='self,sm,ud' export KMP_STACKSIZE=512m # standard version srun ~/qe-7.2/bin/pw.x -i pwscf.in | tee result.out
MPI並列とOpenMP並列のいい塩梅を知っている方いれば、教えてくださると助かります。
タスクあたりのコア数(OpenMP並列)をCPUの構成から見て64の約数にした方がなんとなく良いと思います。
物性研(System B Ohtaka)でのVTST-VASPコンパイル
内輪用です!(重要!)
下準備
まずはmodule loadから
module purge module load oneapi_compiler/2023.0.0 oneapi_mkl/2023.0.0 oneapi_mpi/2023.0.0
次にHDF5のインストール
HDF5® Source Code - The HDF Group
ここからソースコードをダウンロードしてください。
ダウンロードできたらHDF5のディレクトリ内で以下を実行します。
prefix=は各々のディレクトリを指定してください。
./configure --prefix=/home/test/hdf5-1.8.18/ --enable-fortran CC=mpicc FC=mpiifort CXX=mpicxx make make install
VASPのコンパイル
ここではVASPのバージョンは6.4.1を使用してます。
-VTSTの準備
ここではvtstcode 6.4を使用します。
Download — Transition State Tools for VASP
ここから入手できます。
解凍するとvtstcode6.4というディレクトリがあるので、その中に入ってるものを使用します。
"vasp.6.4.1/src"内にmain.Fというファイルがあるのでそれを開いて以下の箇所を次のように書き換えます。
CALL CHAIN_FORCE(T_INFO%NIONS,DYN%POSION,TOTEN,TIFOR, &
LATT_CUR%A,LATT_CUR%B,IO%IU6)
この箇所はこんな感じに
CALL CHAIN_FORCE(T_INFO%NIONS,DYN%POSION,TOTEN,TIFOR, &
TSIF,LATT_CUR%A,LATT_CUR%B,IO%IU6)
IF (LCHAIN) CALL chain_init( T_INFO, IO)
この箇所はこんな感じに
CALL chain_init( T_INFO, IO)
そうしたら"vtstcode6.4"内にあるすべてのファイル/ディレクトリを"vasp.6.4.1/src"内にコピーします。
その際chain.Fが置き換えられるので、バックアップ取るなりなんなりしておいてください。
次に".objects"ファイルを編集します。"vasp.6.4.1/src"内にあります。
隠しファイルなので、なんとかして頑張って開いてください。
ファイルを見てみると
~~~ hamil_rot.o \ chain.o \ ~~~
という箇所があるので、これを以下のように追記します。
~~~ hamil_rot.o \ bfgs.o dynmat.o instanton.o lbfgs.o sd.o cg.o dimer.o bbm.o \ fire.o lanczos.o neb.o qm.o \ pyamff_fortran/*.o ml_pyamff.o \ opt.o chain.o \ ~~~
次に"vasp.6.4.1/src"内にある"makefile"を編集します。
適宜検索して、以下のように変更してください。
LIB= lib parser pyamff_fortran
dependencies: sources libs
これでVTSTの準備は完了です。
-makefile.includeの準備
VASPのコンパイル済みバイナリの利用について - ISSP Supercomputer Center
ここに書いてある通り、"/home/issp/vasp/makefile.include"内に各バージョンごとにmakefile.includeが置いてあるので 基本的にはそれを元にして編集していきます。
CPP_OPTIONSはDFT-D4とかを使用したい場合はVASP wikiを見て書き換えてください。
HDF5_ROOTを下準備でインストールした、HDF5を指定するように書き換えてあげてください。
実行速度を比べてことはないのでなんとも言えないですが、OFLAGを-O3に書き換えたりはあんまりしない方がいいかも?
make
以下のような"~~.sh"ファイルを作ってsbatchでjobを投げればmakeできるはずです。
#!/bin/bash
#SBATCH -J test
#SBATCH -p i8cpu
#SBATCH -N 1
cd ${SLURM_SUBMIT_DIR}
ulimit -s unlimited
module purge
module load oneapi_compiler/2023.0.0 oneapi_mkl/2023.0.0 oneapi_mpi/2023.0.0
export PATH=$PATH:/HDF5をインストールしたディレクトリ
export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:/HDF5をインストールしたディレクトリ/lib
echo $PATH
make DEPS=1 -j 128 std
make時のログはslurm-"jobnumber".outに出力されてるはずです。
VASPKITを使ったUnfoldingしたバンド構造の書き出し方
自分の論文が出版されたので、布教も兼ねて使った手法の解説でもしたいと思います。
https://pubs.acs.org/doi/10.1021/acs.jpcc.2c06048
まずはVASPKITをインストールします。
これから先はVASPKITのTutorialsに書いてあることとほぼ同じなので、適宜そちらも見てください。
まずは真空層を含めた単位セルを作ります。

これは論文中にもあるmonoclinic - ZrO2 (111)面の単位セルですが
注意することがあって、必ずセルの中心が構造(いわゆるスラブ)の中心である必要があります。
(単純に真空層追加するだけじゃダメってこと)
あと酸素欠陥とかはまだ追加しないでね。
次にVASPKITを起動してKPATH.inファイルを作成します。
(必ずさっき作った単位セルで作成すること!)
今回は吸着系を扱うので2D系と同じ方法で以下のように302と打ち込んで作成します。
vaspkit 302
次にスーパーセルを作成します。
今回は2*2*1のもので計算するのでTRANSMAT.inファイルを以下のように作成します。
Read transformation matrix from the TRANSMAT.in file if it exists. 2 0 0 0 2 0 0 0 1
そしたら以下のコマンドを実行します。
vaspkit -task 400
そうするとSUPERCELL.vaspファイルができると思うので、これに適宜酸素欠陥や吸着種を追加していきます。
次に最初に作成したKPATH.inをもとにKPOINTSファイルを作成していきます。
詳細はvaspkitのtutorialsを見てね。
でKPOINTSが用意できたらINCARとPOTCARを用意します。
INCARは各々設定したいパラメーターはあると思うのでアレですが
LWAVE = .TRUE.
だけは必ず設定してね。
重たいWAVECARと共にVASPのJobが終わったら以下を実行します。
vaspkit -task 282
そうするとEBS.datファイルが書き出されると思うので
~/vaspkit/examples/band_unfolding/
の中にあるpythonスクリプトを使ってバンド構造を書き出します。
ちなみに丸の大きさはDOSでいうarb. unitの大きさみたいなものです。
最後に書くのもアレだけど構造最適化はGamma点だけであらかじめ回して
もう一度Slabの中心がスーパーセルの中心に来るようにしてバンド計算を一点計算で回す方が効率的かも。
(この時Slab+molの中心がスーパーセルの中心に来るのではないことは注意)
"Error EDDDAV: Call to ZHEGV failed. Returncode = xx"の解決方法
ここにたどり着いた皆様、ご愁傷様です。
あと多分役に立ちません。すみません。
VASPで出ると一番嫌なエラーである表題を経験則から解決する方法をここでは書いていきます。
他にもあればコメント欄にでもお願いします。
0. 前提
DOSとかBand出す時の一点計算でこのエラーが出るときは明らかに構造がおかしいことが
多いので再度構造最適化をやり直すことを薦めます。
1. ALGO を変える
NormalからFastにするとたまに上手くいく。
Fastの時のエネルギーが正確じゃないかもしれないとかいうのは知りません。
2.PREC を変える
Normal, Med, Accurateあたりを変えてみましょう。HighはENCUTの値を
1.2倍にするだけだからあんまし意味ないと思う。
3. ISTART, ICHARGとかを0にする。
たまに上手くいく、何故かは知らん。
4.(構造最適化のみ) 構造最適化アルゴリズム, POTIM等を変えてみる。
IBRIONもしくは、IOPTの値を変えてみましょう。
ただしそれぞれの構造最適化アルゴリズムについて理解はしておきましょう。
安定構造からかけ離れてるのにIOPT = 7はダメです。
5.他にミスはないですか?
例えば+Uを違う原子に入れてたり、MAGMOMの値を間違ってたり、もう一度INCARを見直しましょう。
6.計算に使用するノード数、CPU数を変えてみる。
何故かは知りませんが、2ノード使用しているとエラー出るのに1ノード使用だとエラーが出ないことがあります。
これは各自PCによりますが、物性研のsystem Bだと64 coreのCPU二つで1ノードなので
shファイルの-Nとか-nの値を弄ると良いかもしれません。
=ここから先はあんまり推奨しない=
7.わざとPOSCARを弄る
その通りに適当などうでも良さそうな原子の座標をちょっと弄ります。
(真空層に加えても良いのかな、やったことないけど...)
それでpolaronの準位ができたり、変な準位ができる可能性があるので構造最適化の時だけにしましょう。
8.IWAVPR, NELDMの値を弄る
prediction of wavefunctions initialized - no I/Oとかmixing delayのエラーが出てる時におすすめ。
どう変えるかは各自調べてください、余白が無いです。
VASP初心者マークの方には絶対にお勧めしません。
9.祈る
闇雲にjobを投げるのはやめましょう。たまには休みましょう。
終わり
Cent OS 7 HDD・SSDマウント方法
計算サーバーにU.2とかいう訳分からん規格のSSDを増設したのでその時のメモ
ホットスワップスロットなりにSSDを突っ込んでサーバーを起動させたら
fdisk -l
で認識されてるのを適当に確認 (U.2だと/dev/nvme0n1p1/って感じで認識されてるはず)
デフォルトで入ってるディスクユーティリティーとかfdisk /dev/~/で出てくる指示に従って適当にパーティション・ファイルシステムを作成する。
そのままでィスクユーティリティー使ってマウントすると、変な場所にマウントされたり再起動する度にマウントしなおさないといけないので
vi /etc/fstab
に書き込みをします。(読み取り用だからsudoでやる)
事前に増設したSSDのUUIDを調べておいて
UUID=(増設したSSDのUUID) /マウントするディレクトリ ext4 defaults 0 0
って感じで追記したら保存したら
mount -a
を必ず実行して再起動(実行しないとメンテナンスモードに入ってしまう)
大抵の計算サーバーはノードがいくつかあってNFSで色々共有してると思うので、そこを弄っていきます。
まず親ノードで
vi /etc/exports
を開いて下みたいな感じで追記
/ノード間で共有したいディレクトリ(マウント先) IPアドレス (rw,no_root_squash,async)
子ノードで
vi /etc/fstab
で下を追記
親ノードIP:/共有ディレクトリ /共有ディレクトリ nfs nfsvers=3,hard,intr,bg,async 0 0
追記したら親ノード・子ノード両方でサービスを再起動
exportfs -ra
で子ノードで
mount -a
これで多分問題なく動きます
一応df -hとか軽いテストジョブを回してちゃんと動くかどうか確認しましょう
分子間相互作用のエネルギーを計算する(psi4・SAPT法)
よく学部の頃の物理化学実験で、何故かわからないことがあったら取り敢えずそいつのせいにしていた分子間相互作用について、計算してみようというやつです。
使うソフト;psi4 (https://psicode.org)
conda使ってインストールするよりinstaller使った方が良い(個人的に)
使う理論;Symmetry-adapted perturbation theory
理論について自分が詳しく説明してもアレなので下のpsi4のサイトやyoutubeを見てください
https://psicode.org/psi4manual/master/sapt.html
www.youtube.com
まぁ要するにElectrostatic(静電)、Exchange(交換)、Induction(誘導)、Dispersion(分散)項に分子間相互作用エネルギーを分解してそれぞれ計算して、それぞれの合算した奴が分子間相互作用エネルギーってことです。
SAPTでのハミルトニアンはどうのこうってのも重要だからちゃんと上のURL見ようね。
で、今回は
“Manipulation” of Crystal Structure by Methylthiolation Enabling Ultrahigh Mobility in a Pyrene-Based Molecular Semiconductor. Adv. Mater. 2021, 33, 2102914. https://doi.org/10.1002/adma.202102914
この論文で行われてるMT-pyreneの計算を"とても曖昧(重要)"に行なって分子間相互作用エネルギーを求めてみます。
まずは適当にGaussViewで分子を作って6-31G(d)程度のレベルで構造最適化します。
し終わったら、根性でそれっぽい位置に分子を置きます。
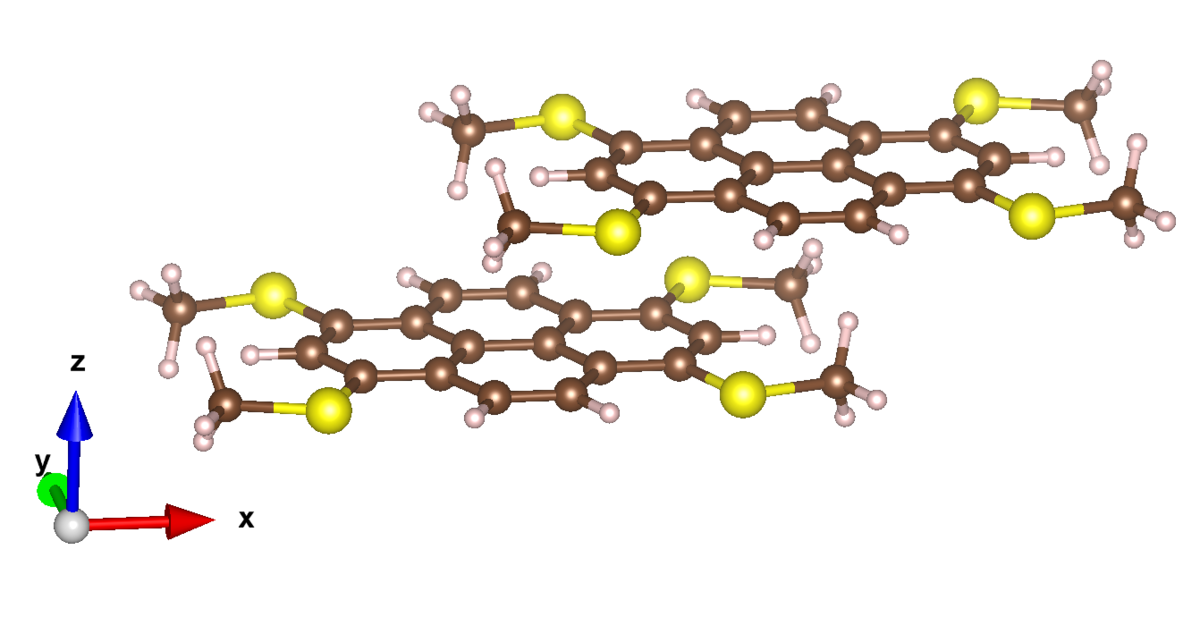
(あくまで曖昧な再現だからそうしてるだけで、実験に即してない第一原理計算は基本的に意味が薄いのでちゃんと実験の測定結果と対照させようね!)
本当は二量体の状態でもGaussianで構造最適化するべきなんだろうけど、面倒くさいのでそのままVESTAなり何なり経由してxyz形式の分子座標ファイルを得てpsi4のinputファイルを作ります。
molecule {
0 1
C -8.623200 -0.057900 -3.147400
C -7.941800 -1.276100 -3.147100
C -6.525100 -1.299300 -3.147300
C -5.814200 -0.057900 -3.147700
C -6.525100 1.183600 -3.147900
C -7.941800 1.160400 -3.147800
C -5.774800 -2.518600 -3.147200
C -4.375000 -0.057900 -3.147800
C -3.664000 -1.299400 -3.147600
C -4.414400 -2.518800 -3.147300
C -2.247000 -1.276300 -3.147700
C -1.565400 -0.057900 -3.148300
C -2.247000 1.160600 -3.148600
C -3.664000 1.183700 -3.148200
C -4.414400 2.403000 -3.148100
C -5.774800 2.402900 -3.148000
H -6.301900 3.351200 -3.147800
H -3.887200 3.351400 -3.147800
H -6.301900 -3.466900 -3.147100
H -9.703300 -0.057900 -3.147300
H -3.887200 -3.467100 -3.147300
H -0.485200 -0.057900 -3.148700
C 0.374900 -2.377600 -3.150300
H 0.921300 -3.324300 -3.150600
H 0.645500 -1.815700 -4.048500
H 0.648700 -1.814500 -2.253900
C 0.374800 2.261900 -3.139800
H 0.921200 3.208500 -3.138100
H 0.641800 1.700600 -2.240200
H 0.652300 1.698200 -4.034800
C -10.563800 2.261500 -3.145800
H -11.110200 3.208100 -3.145600
H -10.837400 1.698500 -4.042400
H -10.834800 1.699500 -2.247800
C -10.563800 -2.377200 -3.148000
H -11.110200 -3.323800 -3.148000
H -10.836900 -1.814300 -2.251200
H -10.835400 -1.815200 -4.045800
S -1.385600 -2.844300 -3.147000
S -1.385600 2.728500 -3.150300
S -8.803300 2.728200 -3.148500
S -8.803300 -2.843900 -3.146400
--
0 1
C -3.528700 0.000000 0.000100
C -2.847300 -1.218300 0.000400
C -1.430600 -1.241500 0.000200
C -0.719800 -0.000000 -0.000200
C -1.430600 1.241500 -0.000400
C -2.847300 1.218300 -0.000300
C -0.680400 -2.460700 0.000300
C 0.719500 -0.000000 -0.000300
C 1.430500 -1.241600 -0.000100
C 0.680100 -2.460900 0.000200
C 2.847500 -1.218500 -0.000200
C 3.529100 -0.000000 -0.000800
C 2.847500 1.218500 -0.001100
C 1.430500 1.241600 -0.000700
C 0.680100 2.460900 -0.000600
C -0.680400 2.460700 -0.000500
H -1.207400 3.409100 -0.000300
H 1.207300 3.409200 -0.000300
H -1.207400 -3.409100 0.000400
H -4.608800 0.000000 0.000200
H 1.207300 -3.409200 0.000200
H 4.609300 -0.000000 -0.001200
C 5.469300 -2.319800 -0.002800
H 6.015700 -3.266400 -0.003100
H 5.740000 -1.757900 -0.901100
H 5.743200 -1.756600 0.893600
C 5.469300 2.319800 0.007700
H 6.015700 3.266400 0.009400
H 5.736300 1.758500 0.907300
H 5.746800 1.756000 -0.887300
C -5.469300 2.319300 0.001700
H -6.015700 3.266000 0.001900
H -5.742900 1.756400 -0.894900
H -5.740400 1.757400 0.899700
C -5.469300 -2.319400 -0.000500
H -6.015700 -3.266000 -0.000500
H -5.742400 -1.756500 0.896300
H -5.740900 -1.757300 -0.898300
S 3.708900 -2.786400 0.000500
S 3.708900 2.786400 -0.002800
S -3.708800 2.786100 -0.001000
S -3.708800 -2.786100 0.001100
units angstrom
}
set {
basis jun-cc-pVDZ
}
energy('sapt0')ちなみにこれだとメモリー使用量はデフォルトのままなので一番上にmemory = 〇〇(任意)Mibとでも追記しときましょう。
でこのインプットを適当なディレクトリに置いて下のコマンドを実行します
psi4 -n X(CPU数) -i (inputファイル名).in -o (outputファイル名).out
そうすると.outファイルがしばらくすると出来るので(12コア仕様で大体1時間)
ファイルを開いて一番最後の方にSAPT resultsってのがあるので、これが分子間相互作用エネルギーになります。
SAPT Results
--------------------------------------------------------------------------------------------------------
Electrostatics -57.22295563 [mEh] -35.90794678 [kcal/mol] -150.23884932 [kJ/mol]
Elst10,r -57.22295563 [mEh] -35.90794678 [kcal/mol] -150.23884932 [kJ/mol]
Exchange 135.81420871 [mEh] 85.22470264 [kcal/mol] 356.58015584 [kJ/mol]
Exch10 135.81420871 [mEh] 85.22470264 [kcal/mol] 356.58015584 [kJ/mol]
Exch10(S^2) 135.25475409 [mEh] 84.87363957 [kcal/mol] 355.11130795 [kJ/mol]
Induction -17.79314427 [mEh] -11.16536660 [kcal/mol] -46.71589385 [kJ/mol]
Ind20,r -62.35322161 [mEh] -39.12723728 [kcal/mol] -163.70836078 [kJ/mol]
Exch-Ind20,r 57.10480365 [mEh] 35.83380529 [kcal/mol] 149.92864132 [kJ/mol]
delta HF,r (2) -12.54472631 [mEh] -7.87193461 [kcal/mol] -32.93617439 [kJ/mol]
Dispersion -94.16843195 [mEh] -59.09158318 [kcal/mol] -247.23918403 [kJ/mol]
Disp20 -111.00924557 [mEh] -69.65935327 [kcal/mol] -291.45473410 [kJ/mol]
Exch-Disp20 16.84081362 [mEh] 10.56777009 [kcal/mol] 44.21555007 [kJ/mol]
Disp20 (SS) -55.50462279 [mEh] -34.82967664 [kcal/mol] -145.72736705 [kJ/mol]
Disp20 (OS) -55.50462279 [mEh] -34.82967664 [kcal/mol] -145.72736705 [kJ/mol]
Exch-Disp20 (SS) 9.68515042 [mEh] 6.07752364 [kcal/mol] 25.42835892 [kJ/mol]
Exch-Disp20 (OS) 7.15566320 [mEh] 4.49024645 [kcal/mol] 18.78719115 [kJ/mol]
Total HF 60.79810880 [mEh] 38.15138926 [kcal/mol] 159.62541267 [kJ/mol]
Total SAPT0 -33.37032315 [mEh] -20.94019392 [kcal/mol] -87.61377136 [kJ/mol]
Special recipe for scaled SAPT0 (see Manual):
Electrostatics sSAPT0 -57.22295563 [mEh] -35.90794678 [kcal/mol] -150.23884932 [kJ/mol]
Exchange sSAPT0 135.81420871 [mEh] 85.22470264 [kcal/mol] 356.58015584 [kJ/mol]
Induction sSAPT0 -17.08160094 [mEh] -10.71886642 [kcal/mol] -44.84773708 [kJ/mol]
Dispersion sSAPT0 -93.95859026 [mEh] -58.95990553 [kcal/mol] -246.68824474 [kJ/mol]
Total sSAPT0 -32.44893812 [mEh] -20.36201609 [kcal/mol] -85.19467530 [kJ/mol]
--------------------------------------------------------------------------------------------------------TotalのSAPTエネルギーは論文中だと–23.38 kcal/molで上の計算だと-20.36 kcal/molでまぁまぁ近い値なんじゃ無いかと!
ただtotal以外のSAPTエネルギーは凄い違いが出てるので多分適当に起きすぎたんだろうな...
まぁ位置なんて言うのはちゃんと構造最適化したり実験で測定したりしたら出るのでまぁ良いとして、大事なのは分子間相互作用って言うけど、
具体的に分子の何と何の相互作用があるのかちゃんと認識して考えて計算することが重要です。(この論文だとMT-Pyreneの場合π面とπ面同士とCH基とCH基同士)
で次にじゃあ色々求めた分子間相互作用のSAPTエネルギーのうち、どれが分子の集合体に対して安定性に影響を与えているのか?とか色々考えられることはあるね!
もしMT-PyreneがMoS2みたいに平面上に並ぶのなら両端のCH同士のSAPTエネルギーの内何かが、π面とπ面同士のFace to FaceのSAPTエネルギーよりも低くなって平面上に広がるとか考えれそう(知らんけど)
こういう有機半導体の計算も固体表面ばっかりやってる身からしたら新鮮で良いね。