分子間相互作用のエネルギーを計算する(psi4・SAPT法)
よく学部の頃の物理化学実験で、何故かわからないことがあったら取り敢えずそいつのせいにしていた分子間相互作用について、計算してみようというやつです。
使うソフト;psi4 (https://psicode.org)
conda使ってインストールするよりinstaller使った方が良い(個人的に)
使う理論;Symmetry-adapted perturbation theory
理論について自分が詳しく説明してもアレなので下のpsi4のサイトやyoutubeを見てください
https://psicode.org/psi4manual/master/sapt.html
www.youtube.com
まぁ要するにElectrostatic(静電)、Exchange(交換)、Induction(誘導)、Dispersion(分散)項に分子間相互作用エネルギーを分解してそれぞれ計算して、それぞれの合算した奴が分子間相互作用エネルギーってことです。
SAPTでのハミルトニアンはどうのこうってのも重要だからちゃんと上のURL見ようね。
で、今回は
“Manipulation” of Crystal Structure by Methylthiolation Enabling Ultrahigh Mobility in a Pyrene-Based Molecular Semiconductor. Adv. Mater. 2021, 33, 2102914. https://doi.org/10.1002/adma.202102914
この論文で行われてるMT-pyreneの計算を"とても曖昧(重要)"に行なって分子間相互作用エネルギーを求めてみます。
まずは適当にGaussViewで分子を作って6-31G(d)程度のレベルで構造最適化します。
し終わったら、根性でそれっぽい位置に分子を置きます。
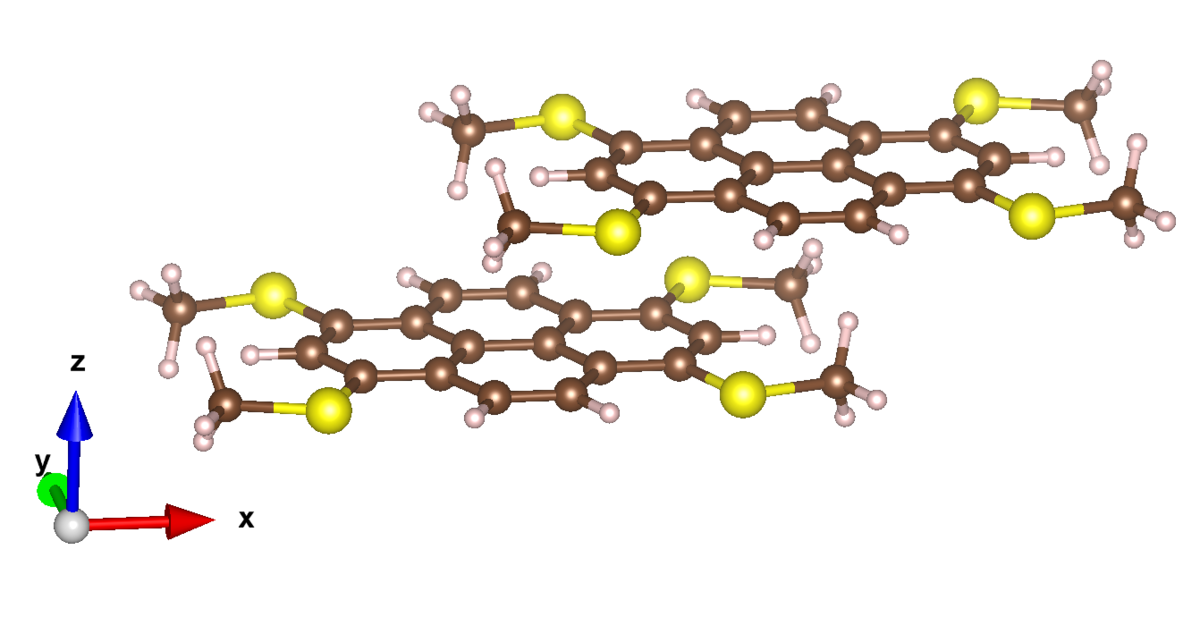
(あくまで曖昧な再現だからそうしてるだけで、実験に即してない第一原理計算は基本的に意味が薄いのでちゃんと実験の測定結果と対照させようね!)
本当は二量体の状態でもGaussianで構造最適化するべきなんだろうけど、面倒くさいのでそのままVESTAなり何なり経由してxyz形式の分子座標ファイルを得てpsi4のinputファイルを作ります。
molecule {
0 1
C -8.623200 -0.057900 -3.147400
C -7.941800 -1.276100 -3.147100
C -6.525100 -1.299300 -3.147300
C -5.814200 -0.057900 -3.147700
C -6.525100 1.183600 -3.147900
C -7.941800 1.160400 -3.147800
C -5.774800 -2.518600 -3.147200
C -4.375000 -0.057900 -3.147800
C -3.664000 -1.299400 -3.147600
C -4.414400 -2.518800 -3.147300
C -2.247000 -1.276300 -3.147700
C -1.565400 -0.057900 -3.148300
C -2.247000 1.160600 -3.148600
C -3.664000 1.183700 -3.148200
C -4.414400 2.403000 -3.148100
C -5.774800 2.402900 -3.148000
H -6.301900 3.351200 -3.147800
H -3.887200 3.351400 -3.147800
H -6.301900 -3.466900 -3.147100
H -9.703300 -0.057900 -3.147300
H -3.887200 -3.467100 -3.147300
H -0.485200 -0.057900 -3.148700
C 0.374900 -2.377600 -3.150300
H 0.921300 -3.324300 -3.150600
H 0.645500 -1.815700 -4.048500
H 0.648700 -1.814500 -2.253900
C 0.374800 2.261900 -3.139800
H 0.921200 3.208500 -3.138100
H 0.641800 1.700600 -2.240200
H 0.652300 1.698200 -4.034800
C -10.563800 2.261500 -3.145800
H -11.110200 3.208100 -3.145600
H -10.837400 1.698500 -4.042400
H -10.834800 1.699500 -2.247800
C -10.563800 -2.377200 -3.148000
H -11.110200 -3.323800 -3.148000
H -10.836900 -1.814300 -2.251200
H -10.835400 -1.815200 -4.045800
S -1.385600 -2.844300 -3.147000
S -1.385600 2.728500 -3.150300
S -8.803300 2.728200 -3.148500
S -8.803300 -2.843900 -3.146400
--
0 1
C -3.528700 0.000000 0.000100
C -2.847300 -1.218300 0.000400
C -1.430600 -1.241500 0.000200
C -0.719800 -0.000000 -0.000200
C -1.430600 1.241500 -0.000400
C -2.847300 1.218300 -0.000300
C -0.680400 -2.460700 0.000300
C 0.719500 -0.000000 -0.000300
C 1.430500 -1.241600 -0.000100
C 0.680100 -2.460900 0.000200
C 2.847500 -1.218500 -0.000200
C 3.529100 -0.000000 -0.000800
C 2.847500 1.218500 -0.001100
C 1.430500 1.241600 -0.000700
C 0.680100 2.460900 -0.000600
C -0.680400 2.460700 -0.000500
H -1.207400 3.409100 -0.000300
H 1.207300 3.409200 -0.000300
H -1.207400 -3.409100 0.000400
H -4.608800 0.000000 0.000200
H 1.207300 -3.409200 0.000200
H 4.609300 -0.000000 -0.001200
C 5.469300 -2.319800 -0.002800
H 6.015700 -3.266400 -0.003100
H 5.740000 -1.757900 -0.901100
H 5.743200 -1.756600 0.893600
C 5.469300 2.319800 0.007700
H 6.015700 3.266400 0.009400
H 5.736300 1.758500 0.907300
H 5.746800 1.756000 -0.887300
C -5.469300 2.319300 0.001700
H -6.015700 3.266000 0.001900
H -5.742900 1.756400 -0.894900
H -5.740400 1.757400 0.899700
C -5.469300 -2.319400 -0.000500
H -6.015700 -3.266000 -0.000500
H -5.742400 -1.756500 0.896300
H -5.740900 -1.757300 -0.898300
S 3.708900 -2.786400 0.000500
S 3.708900 2.786400 -0.002800
S -3.708800 2.786100 -0.001000
S -3.708800 -2.786100 0.001100
units angstrom
}
set {
basis jun-cc-pVDZ
}
energy('sapt0')ちなみにこれだとメモリー使用量はデフォルトのままなので一番上にmemory = 〇〇(任意)Mibとでも追記しときましょう。
でこのインプットを適当なディレクトリに置いて下のコマンドを実行します
psi4 -n X(CPU数) -i (inputファイル名).in -o (outputファイル名).out
そうすると.outファイルがしばらくすると出来るので(12コア仕様で大体1時間)
ファイルを開いて一番最後の方にSAPT resultsってのがあるので、これが分子間相互作用エネルギーになります。
SAPT Results
--------------------------------------------------------------------------------------------------------
Electrostatics -57.22295563 [mEh] -35.90794678 [kcal/mol] -150.23884932 [kJ/mol]
Elst10,r -57.22295563 [mEh] -35.90794678 [kcal/mol] -150.23884932 [kJ/mol]
Exchange 135.81420871 [mEh] 85.22470264 [kcal/mol] 356.58015584 [kJ/mol]
Exch10 135.81420871 [mEh] 85.22470264 [kcal/mol] 356.58015584 [kJ/mol]
Exch10(S^2) 135.25475409 [mEh] 84.87363957 [kcal/mol] 355.11130795 [kJ/mol]
Induction -17.79314427 [mEh] -11.16536660 [kcal/mol] -46.71589385 [kJ/mol]
Ind20,r -62.35322161 [mEh] -39.12723728 [kcal/mol] -163.70836078 [kJ/mol]
Exch-Ind20,r 57.10480365 [mEh] 35.83380529 [kcal/mol] 149.92864132 [kJ/mol]
delta HF,r (2) -12.54472631 [mEh] -7.87193461 [kcal/mol] -32.93617439 [kJ/mol]
Dispersion -94.16843195 [mEh] -59.09158318 [kcal/mol] -247.23918403 [kJ/mol]
Disp20 -111.00924557 [mEh] -69.65935327 [kcal/mol] -291.45473410 [kJ/mol]
Exch-Disp20 16.84081362 [mEh] 10.56777009 [kcal/mol] 44.21555007 [kJ/mol]
Disp20 (SS) -55.50462279 [mEh] -34.82967664 [kcal/mol] -145.72736705 [kJ/mol]
Disp20 (OS) -55.50462279 [mEh] -34.82967664 [kcal/mol] -145.72736705 [kJ/mol]
Exch-Disp20 (SS) 9.68515042 [mEh] 6.07752364 [kcal/mol] 25.42835892 [kJ/mol]
Exch-Disp20 (OS) 7.15566320 [mEh] 4.49024645 [kcal/mol] 18.78719115 [kJ/mol]
Total HF 60.79810880 [mEh] 38.15138926 [kcal/mol] 159.62541267 [kJ/mol]
Total SAPT0 -33.37032315 [mEh] -20.94019392 [kcal/mol] -87.61377136 [kJ/mol]
Special recipe for scaled SAPT0 (see Manual):
Electrostatics sSAPT0 -57.22295563 [mEh] -35.90794678 [kcal/mol] -150.23884932 [kJ/mol]
Exchange sSAPT0 135.81420871 [mEh] 85.22470264 [kcal/mol] 356.58015584 [kJ/mol]
Induction sSAPT0 -17.08160094 [mEh] -10.71886642 [kcal/mol] -44.84773708 [kJ/mol]
Dispersion sSAPT0 -93.95859026 [mEh] -58.95990553 [kcal/mol] -246.68824474 [kJ/mol]
Total sSAPT0 -32.44893812 [mEh] -20.36201609 [kcal/mol] -85.19467530 [kJ/mol]
--------------------------------------------------------------------------------------------------------TotalのSAPTエネルギーは論文中だと–23.38 kcal/molで上の計算だと-20.36 kcal/molでまぁまぁ近い値なんじゃ無いかと!
ただtotal以外のSAPTエネルギーは凄い違いが出てるので多分適当に起きすぎたんだろうな...
まぁ位置なんて言うのはちゃんと構造最適化したり実験で測定したりしたら出るのでまぁ良いとして、大事なのは分子間相互作用って言うけど、
具体的に分子の何と何の相互作用があるのかちゃんと認識して考えて計算することが重要です。(この論文だとMT-Pyreneの場合π面とπ面同士とCH基とCH基同士)
で次にじゃあ色々求めた分子間相互作用のSAPTエネルギーのうち、どれが分子の集合体に対して安定性に影響を与えているのか?とか色々考えられることはあるね!
もしMT-PyreneがMoS2みたいに平面上に並ぶのなら両端のCH同士のSAPTエネルギーの内何かが、π面とπ面同士のFace to FaceのSAPTエネルギーよりも低くなって平面上に広がるとか考えれそう(知らんけど)
こういう有機半導体の計算も固体表面ばっかりやってる身からしたら新鮮で良いね。